使用 clusterProfiler 的内置富集函数做代谢物富集分析。
# 加载必要的包
library(clusterProfiler)
##
## Registered S3 methods overwritten by 'treeio':
## method from
## MRCA.phylo tidytree
## MRCA.treedata tidytree
## Nnode.treedata tidytree
## Ntip.treedata tidytree
## ancestor.phylo tidytree
## ancestor.treedata tidytree
## child.phylo tidytree
## child.treedata tidytree
## full_join.phylo tidytree
## full_join.treedata tidytree
## groupClade.phylo tidytree
## groupClade.treedata tidytree
## groupOTU.phylo tidytree
## groupOTU.treedata tidytree
## inner_join.phylo tidytree
## inner_join.treedata tidytree
## is.rooted.treedata tidytree
## nodeid.phylo tidytree
## nodeid.treedata tidytree
## nodelab.phylo tidytree
## nodelab.treedata tidytree
## offspring.phylo tidytree
## offspring.treedata tidytree
## parent.phylo tidytree
## parent.treedata tidytree
## root.treedata tidytree
## rootnode.phylo tidytree
## sibling.phylo tidytree
## clusterProfiler v4.10.0 For help: https://yulab-smu.top/biomedical-knowledge-mining-book/
##
## If you use clusterProfiler in published research, please cite:
## T Wu, E Hu, S Xu, M Chen, P Guo, Z Dai, T Feng, L Zhou, W Tang, L Zhan, X Fu, S Liu, X Bo, and G Yu. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation. 2021, 2(3):100141
##
## Attaching package: 'clusterProfiler'
## The following object is masked from 'package:stats':
##
## filter
# 构造感兴趣的代谢物列表
metabolite_list = c("Metabolite1", "Metabolite2", "Metabolite3", "Metabolite6", "Metabolite7")
# 构造代谢途径与代谢物的映射关系 (TERM2GENE)
term2gene = data.frame(
Pathway = c("Pathway1", "Pathway1", "Pathway1", "Pathway2", "Pathway2",
"Pathway3", "Pathway3", "Pathway4", "Pathway4", "Pathway5"),
Metabolite = c("Metabolite1", "Metabolite2", "Metabolite3", "Metabolite4", "Metabolite5",
"Metabolite6", "Metabolite7", "Metabolite8", "Metabolite9", "Metabolite10")
)
# 构造代谢途径描述信息 (TERM2NAME)
term2name = data.frame(
Pathway = c("Pathway1", "Pathway2", "Pathway3", "Pathway4", "Pathway5"),
Description = c("Metabolic Pathway 1", "Metabolic Pathway 2", "Metabolic Pathway 3",
"Metabolic Pathway 4", "Metabolic Pathway 5")
)
# 使用 enricher 函数进行代谢物富集分析
result = enricher(
gene = metabolite_list,
pvalueCutoff = 0.5, # 调高 cutoff 以得到结果
pAdjustMethod = "BH",
TERM2GENE = term2gene,
TERM2NAME = term2name,
minGSSize = 1 # 将默认的 10 改成 1,否则也得不到结果
)
# 查看结果
if (!is.null(result)) {
print(head(as.data.frame(result)))
# 可视化结果
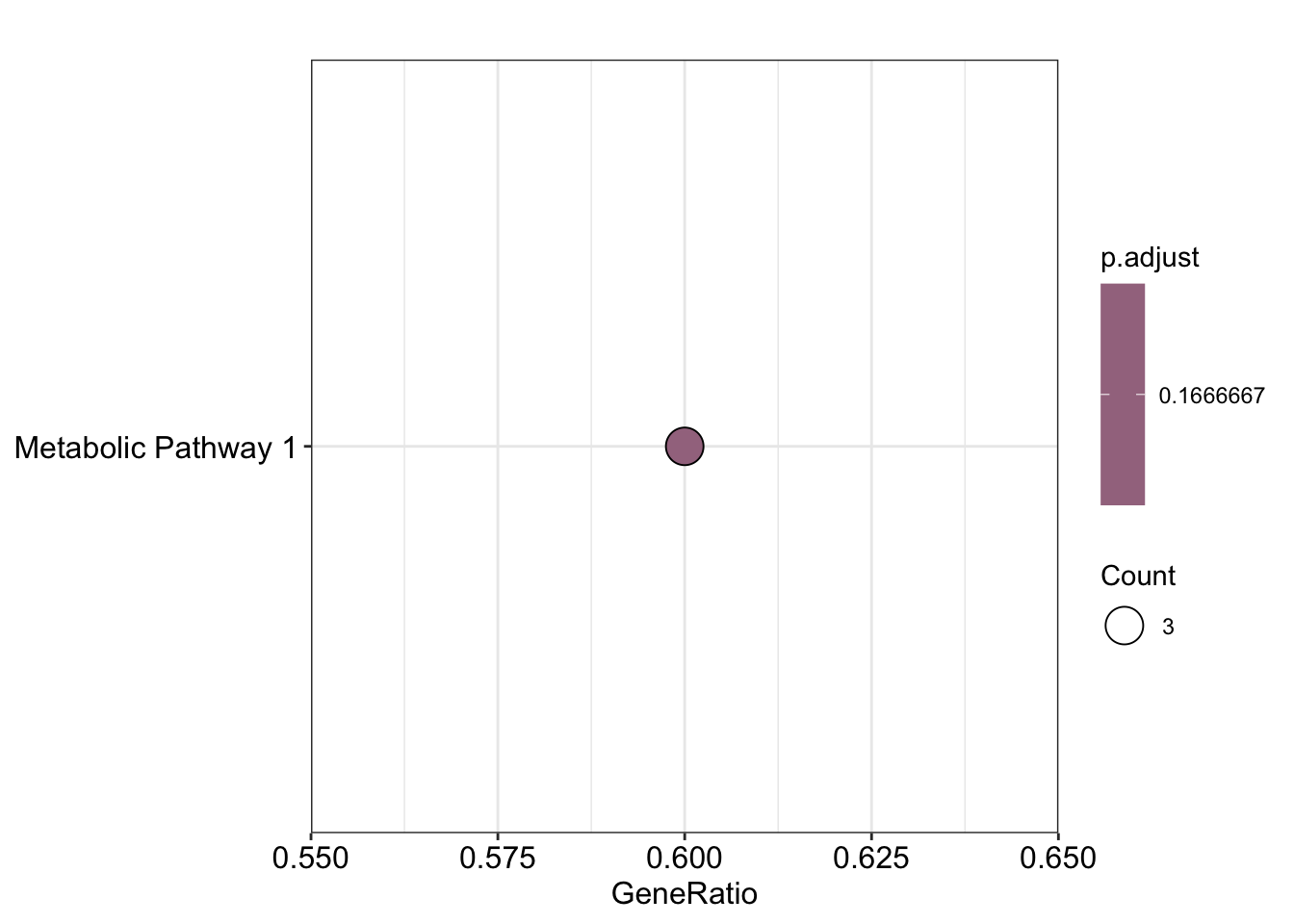
dotplot(result, showCategory = 5)
} else {
message("没有显著富集的代谢途径")
}
## ID Description GeneRatio BgRatio pvalue p.adjust
## Pathway1 Pathway1 Metabolic Pathway 1 3/5 3/10 0.08333333 0.1666667
## qvalue geneID Count
## Pathway1 0.1666667 Metabolite1/Metabolite2/Metabolite3 3